 第七章 体内药物分析法 第七章 体内药物分析法 |
||||||||||||||||||
|
|
||||||||||||||||||
考试要求:
|
||||||||||||||||||
药物的体内过程: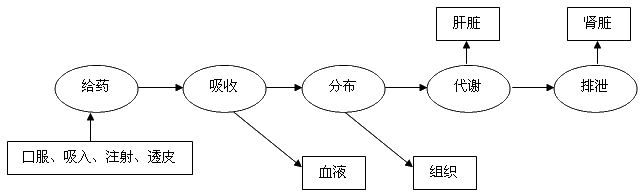 治疗药物监测:以灵敏可靠的方法,检测病人在给药后的血液或其他体液中的药物浓度,并应用药物代谢动力学理论,指导最适个体化用药方案的制定和调整,以避免用药剂量过大及可能产生的毒性反应,保证药物治疗的有效性和安全性。 |
||||||||||||||||||
 |
||||||||||||||||||
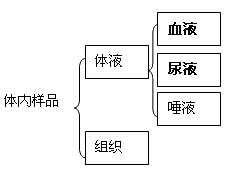
体内样品的采集时间
|
||||||||||||||||||
一、血样 |
||||||||||||||||||
|
血清、血浆的区别 加入抗凝剂后,全血离心制的血浆。不加抗凝剂,自然沉降后离心制的血清。 采血后应尽快(一般不超过2h )分离出血浆、血清,再冰冻保存(-20℃)。 |
||||||||||||||||||
|
二、尿液 用于药物剂量回收、药物肾清除率及生物利用度的研究,以及药物代谢物及其代谢途径、类型和速率等的研究。 PH:4.8-8.0 1、尿液的分类  |
||||||||||||||||||
|
2、尿液的采集 测定尿液中药物浓度——自然排尿、时间尿 测定尿液中药物总量——自然排尿、各时间段全部尿液 3、尿液的处理方法 保存:低温保存,或加入防腐剂。 处理:酸、酶水解 |
||||||||||||||||||
三、唾液 PH:6.2-7.6 1、唾液的采集 漱口后15min。 2、唾液的处理方法 保存:冷藏 处理:去除泡沫,离心取上清液,加碱处理(分离黏蛋白) |
||||||||||||||||||
|
一、体内样品处理的目的 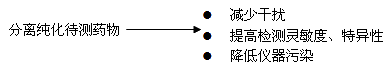 二、体内样品处理的常用方法 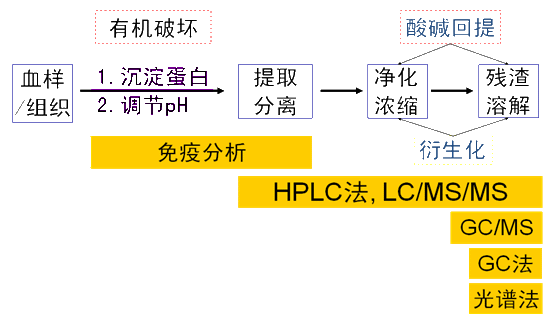 |
||||||||||||||||||
|
1、去除蛋白质法 去除蛋白质的意义——使结合型药物释放出来,得到较“干净”的提取液,消除对测定的干扰 去除蛋白质的方法: 蛋白质沉淀法 加与水混溶的有机溶剂脱水 加入强酸生成不溶性盐 加无机盐盐析 蛋白分解法——蛋白水解酶(枯草菌溶素) |
||||||||||||||||||
|
2、缀合物水解法 缀合物水解的意义——药物经二相代谢可以形成葡糖醛酸苷或硫酸酯缀合物,为了准确测定体内样品中药物的含量,首先需将缀合物水解释放出缀合的药物或其代谢物后,再进行进一步的处理测定。 缀合物水解的方法: 酸水解(HCl):简便、快速,但专一性差 酶水解:β-葡萄糖醛酸苷酶、硫酸酯酶、两者混合酶,专属性高 3、分离纯化与浓集法 分离、纯化、浓集的意义——提高分析的特异性和灵敏度。 常用方法: 液-液萃取——选用与水不相混溶的有机溶剂进行提取 溶剂提取的关键问题是——提取的选择性和提取率的重复性 影响提取率的因素主要有: 水相pH、提取溶剂、离子强度 离子对萃取法 固相萃取法——利用柱色谱进行分离 |
||||||||||||||||||
|
固相萃取法 利用柱色谱分离方法,将具有吸附、分配或离子交换性质的大比表面积吸附填料作为萃取剂填成小柱,以适宜溶剂淋洗活化后,将体内样品通过小柱,微量的待测成分被保留,而内源性干扰物质则大都不被保留而通过,用淋洗溶剂进一步洗去干扰物后,再用洗脱溶剂将药物洗脱收集,挥干溶剂后,用少量溶剂复溶进行分析。 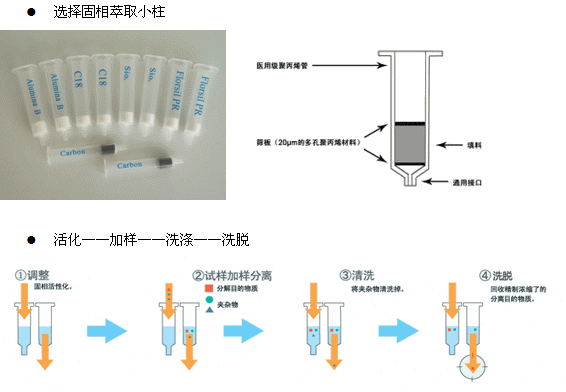 |
||||||||||||||||||
|
4、化学衍生化法 ①衍生化目的 增加组分的稳定性 改善被测组分的色谱行为 增加被测物的检测灵敏度和选择性 提高对光学异构体分离的能力 ②衍生化方法 GC——硅烷化、酰化、烷基化 HPLC——荧胺、丹酰氯、邻苯二醛(柱前衍生化、柱后衍生化) 手性衍生化法 |
||||||||||||||||||
|
一、常用测定方法
|
||||||||||||||||||
|
二、定量分析方验证 1、特异性 内源性物质的干扰、代谢产物的干扰、同用药物的干扰 要求:6个不同来源的空白体内样品色谱图、空白体内样品外加标准物质色谱图(注明浓度)及用药后的体内样品色谱图。 (体外分析的专属性) 2、标准曲线与线性范围 要求:用至少6个浓度建立标准曲线,应使用与待测样品相同的生物介质,线性范围要能覆盖全部待测浓度,不允许将线性范围外推求算未知样品的浓度。随行空白体内样品,但标准曲线不包括零点。(体外分析要求至少5个浓度) |
||||||||||||||||||
|
3、定量下限 标准曲线上的最低浓度点。S/N应大于5。实际测定时,满足3~5个半衰期浓度,或Cmax1/10~1/20。至少5个标准样品测试结果证明。 (体外分析要求S/N=10) 4、精密度与准确度 样品选择:选择3个浓度的质控样品同时进行方法的精密度和准确度考察。低浓度选择在定量下限3倍以内,高浓度接近于标准曲线的上限附近;中间选一个浓度。每一浓度每批至少测定5个样品。 精密度:批内精密度、批间精密度(不同天连续制备3个分析批)。用RSD表示,要求小于15%,在最低定量限附近小于20%。 准确度:准确度用相对回收率表示。一般应在85%~115%范围内,在最低定量限附近应在80%~120%范围内。(体外分析要求95-105%) |
||||||||||||||||||
|
5、样品稳定性 表示一种分析物在确定条件下,一定时间内在给定介质中的化学稳定性。 要求:根据具体情况,对含药生物样品(或储备液、样品处理液)在室温、冰冻或冻融条件下以及不同存放时间进行稳定性考察。 6、提取回收率 分析过程的提取效率,能反映出样品预处理过程中组分丢失的情况,是评价萃取方案优劣的指标之一。以样品提取和处理过程前后分析物含量百分比表示。 要求:考察高、中、低3个浓度的提取回收率,其值一般低于100%(可能低至60%),但重要的是重现性好。 |
||||||||||||||||||
|
7、质控样品 在生物介质中加入已知量待测药物所配制的样品,用于质量控制。 8、质量控制 应在生物样品分析方法确证完成之后开始测试未知样品,每个未知样品一般测定一次,必要时可进行复测。 每批生物样品测定时应建立新的标准曲线,并平行测定高、中、低3个浓度的质控样品。 质控样品测定结果的偏差一般应小于15%。 9、测试结果 |
||||||||||||||||||
|
三、应用 治疗药物监测——易中毒的药物 药代动力学研究 盐酸吗啡、磷酸可待因、硫酸庆大霉素体内、阿奇霉素 历年考点 体内样品的处理方法 |
||||||||||||||||||
|
||||||||||||||||||










